This search tool takes a FASTA file containing up to 20 nucleotide sequences, each with length no greater than 10,000 bp, and scans each sequence for putative transcriptoin factor binding sites. The routine identifies putative binding sites using an 8 bp sliding window, where, for each window sequence, any factor whose corresponding 8-mer enrichment score exceeds the enrichment score threshold (default 0.45) is considered a positive hit. The results of this scan are reported both in an HTML table, which can be downloaded as plain text, and in a graph similar to the UCSC Genome Browser's "dense" view of annotations, where each factor with at least one putative binding site is represented as a track.
Please do not use 'N' or 'X' in the sequences; 'N' and 'X' nucleotides are not yet supported.
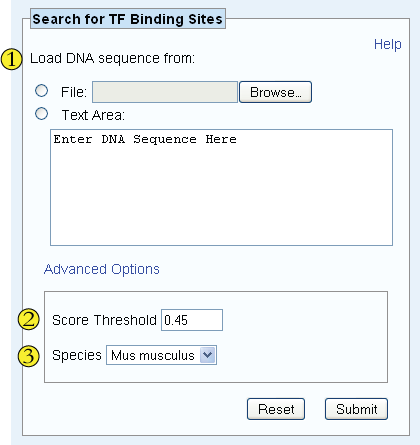
 Submit the FASTA data either by uploading a file or by insering the text directly into the Text Area. As an alternative to a FASTA file, the user may provide a file (or text in the Text Area) where each line contains a single nucleotide sequence. Upon submission, the tool will then label the input as sequences 1 through n, where n is the number of lines entered.
Submit the FASTA data either by uploading a file or by insering the text directly into the Text Area. As an alternative to a FASTA file, the user may provide a file (or text in the Text Area) where each line contains a single nucleotide sequence. Upon submission, the tool will then label the input as sequences 1 through n, where n is the number of lines entered.
 Enrichment scores are calculated using a variant of the Wilcoxon-Mann-Whitney statistic and range from 0.5 for most favored k-mers to -0.5 for most disfavored k-mers. The Score Threshold denotes the minimum enrichment score that an 8-mer for a particular factor must have in order to be considered a putative binding factor. By default, this value is set to 0.45, a score that we consider highly significant for most factor k-mers.
Enrichment scores are calculated using a variant of the Wilcoxon-Mann-Whitney statistic and range from 0.5 for most favored k-mers to -0.5 for most disfavored k-mers. The Score Threshold denotes the minimum enrichment score that an 8-mer for a particular factor must have in order to be considered a putative binding factor. By default, this value is set to 0.45, a score that we consider highly significant for most factor k-mers.
 Specify the species from which candidate transcription factors should be chosen.
Specify the species from which candidate transcription factors should be chosen.