

Cis Regulatory Codes
Functional annotation of DNA regulatory motifs (typically ~6-15 bp in length) is fundamentally important for understanding transcriptional regulatory networks. Effective computational methods for mapping DNA regulatory motifs exist in the yeast Saccharomyces cerevisiae, where the DNA binding sites of regulatory transcription factors (TFs) typically occur within ~600 bp upstream of genes. However, these methods cannot be applied to metazoan genomes, where genes in the same expression cluster are not necessarily co-regulated by a common mechanism, and the regulatory elements can be far from the transcription start site.
 In metazoans, regulatory motifs tend to co-occur within stretches of noncoding sequence, referred to as cis regulatory modules (CRMs), that regulate expression of the nearby gene(s).
Numerous approaches have resulted in the successful identification of CRMs, but such approaches do not attempt to predict ab initio the gene expression patterns or functions
of the genes regulated by the CRMs. Although algorithms have been developed recently for evaluating the regulatory significance of CRM binding site composition, thus far they have
been unable to evaluate the vast sequence regions beyond the proximal promoter that must be considered in mammalian genomes.
In metazoans, regulatory motifs tend to co-occur within stretches of noncoding sequence, referred to as cis regulatory modules (CRMs), that regulate expression of the nearby gene(s).
Numerous approaches have resulted in the successful identification of CRMs, but such approaches do not attempt to predict ab initio the gene expression patterns or functions
of the genes regulated by the CRMs. Although algorithms have been developed recently for evaluating the regulatory significance of CRM binding site composition, thus far they have
been unable to evaluate the vast sequence regions beyond the proximal promoter that must be considered in mammalian genomes.
Because of these complications, analyses of transcriptional regulatory elements in mammals have focused either on the prediction of CRMs starting with a collection of known co-regulatory TFs whose DNA binding specificities are available and a set of genes that the TFs may regulate, or on the computational identification of "motif dictionaries." However, with the advent of high-throughput methods for assembling motif dictionaries, from either chromatin immunoprecipitations or protein binding microarrays, the major computational problem to solve will shift from motif prediction to identifying and associating CRMs to both specific genes and biological processes.
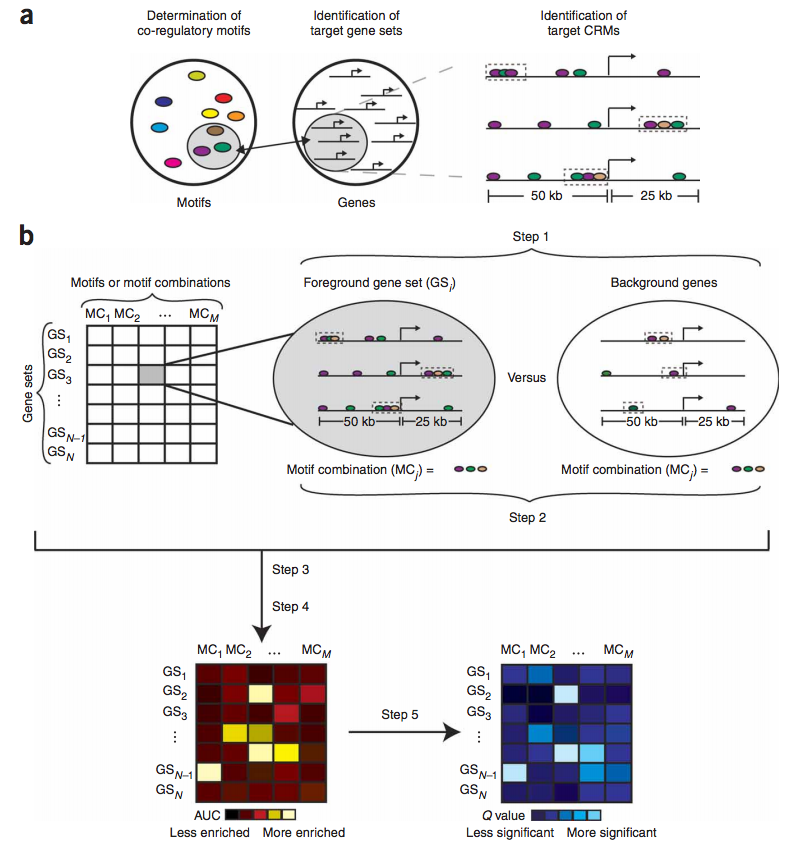
Therefore, we have developed a computational algorithm (termed "Lever") that systematically identifies the target gene sets that are likely to be regulated by a query collection of candidate regulatory motifs (Warner, Philippakis, Jaeger et al., Nature Methods. 2008, 5(4):347-53). Candidate regulatory motifs can come from various sources, including the literature, databases, comparative genomics, protein binding microarrays, and ChIP-chip or ChIP-seq. The ability to screen many gene sets with many motifs /motif combinations allows us to tackle the difficulty in a priori identification of co-regulated gene sets. Lever does not perform de novo motif discovery, but rather evaluates an input collection of motifs for enrichment within candidate CRMs in the noncoding sequences flanking various input gene sets. Lever accomplishes this by assessing whether the motifs are enriched within CRMs, predicted by our PhylCRM (pronounced fulcrum) algorithm, in the noncoding sequences surrounding genes in a collection of gene sets. When these gene sets correspond to Gene Ontology (GO) categories, the results of Lever analysis allow the unbiased assignment of functional annotations to the regulatory motifs and also to the candidate CRMs that comprise the genomic motif occurrences.
We have applied these algorithms to various systems, both in mammals and Drosophila. Using human myogenic differentiation as a model system, we statistically assessed greater than 25,000 pairings of gene sets and motifs / motif combinations. These results allowed us to assign functional annotations to candidate regulatory motifs predicted previously, to identify gene sets that are likely to be co-regulated via shared regulatory motifs, and to identify novel, functional transcriptional enhancers. We have performed similar analyses and experiments in Drosophila for various embryonic cell types important in the development of somatic and cardiac mesoderm.
Lever allows moving beyond the identification of candidate regulatory motifs in mammalian genomes, towards understanding their biological roles. This method represents a major genome-wide step in moving from a motif dictionary to understanding the language of cis regulation. This approach is general and can be applied readily to any cell type, gene expression pattern, or organism of interest.