

Cis Regulatory Elements
As described on the Cis Regulatory Codes page, transcriptional regulation in metazoans is complex and regulatory elements can be far from a gene's transcription start site. In metazoans, gene expression is regulated in a tissue/cell-type-specific manner predominantly via stretches of noncoding sequence referred to as cis regulatory modules (CRMs). CRMs contain 1 or more DNA binding sites for 1 or more sequence-specific, regulatory transcription factors that function to modulate the expression of target gene(s). CRMs that activate gene expression are typically referred to as enhancers while those that repress gene expression are referred to as silencers. Transcriptional enhancers activate gene expression in a tissue-specific manner in development and also in adult cells in response to cellular or environmental stimuli.
We have developed computational algorithms that predict CRMs in the noncoding sequences flanking genes of interest. We previously developed an algorithm called PhylCRM (Warner, Philippakis, Jaeger et al., Nature Methods. 2008, 5(4):347-353), which combines data for individual motif occurrences scored on an alignment into a single CRM prediction. PhylCRM can scan very long genomic sequences for candidate CRMs by quantifying both motif clustering and conservation across arbitrarily many genomes using an evolutionary model consistent with the phylogeny of the genomes. In our study of cis regulatory elements involved in human myogenic differentiation, we examined 75 kb around the transcription start sites of genes, and utilized the phylogenetic tree containing all 8 sequenced mammalian genomes (human, chimp, macaque, mouse, rat, dog, cow, and opossum). Significantly scoring candidate CRMs of varying lengths, ranging from 20 to 500 bp, were identified and scored.
We have applied these approaches to various systems, both in mammals and Drosophila. The results allowed us to successfully identify novel, functional transcriptional enhancers. This approach is general and can be applied readily to any metazoan genome of interest.
Even though transcriptional enhancers have been the focus of many computational and experimental studies, identification of tissue/cell-type-specific enhancers in metazoans remains a significant challenge.
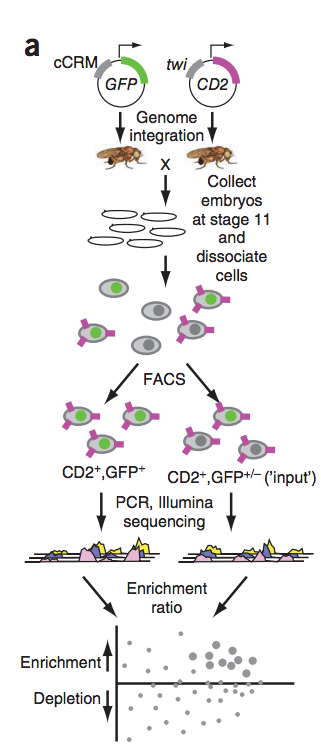
Previously, we developed 'enhancer-FACS-Seq' (eFS) technology (see figure to the left) for highly parallel identification of active, tissue-specific enhancers in Drosophila embryos. Analysis of enhancers identified by eFS as being active in mesodermal tissues revealed enriched DNA binding site motifs of known and putative, novel mesodermal transcription factors (TFs). Naïve Bayes classifiers using TF binding site motifs accurately predicted mesodermal enhancer activity. Application of eFS and related technologies to other cell types and organisms should accelerate the cataloging of enhancers and understanding how transcriptional regulation is encoded within them.
Epistatic interactions among cis-regulatory motifs
We recently developed Quantitative enhancer-FACS-Seq (QeFS) technology for highly parallel quantification of enhancer activities from a common chromosomal locus in Drosophila melanogaster embryos (Waters et al., submitted). Using QeFS, we investigated the contributions of the DNA binding motifs of four poorly characterized TFs to the activities of twelve embryonic mesodermal enhancers across a panel of wild type and mutant enhancers. By measuring the effects of motif mutations both individually and in combination with each other on enhancer activity, we discovered a range of epistatic interactions among the motifs, including both synergistic and alleviating interactions. Altogether, our results indicate that understanding the regulatory consequences of TF binding motifs requires that they be investigated not just individually but also in combination with each other, across a panel of enhancers. Elucidating the context-dependent interplay between TF binding motifs will be important for understanding how cis-regulatory information is encoded in transcriptional enhancers.